The 3Rs of the genome: Reading, writing and regulating
Posted on March 10, 2021UNIVERSITY PARK, Pa. — A massive effort to map the precise binding locations of over 400 different kinds of proteins on the yeast genome has produced the most thorough and high-resolution map of chromosome architecture and gene regulation to date.
The study reveals two distinct gene regulatory architectures, expanding the traditional model of gene regulation. So-called constitutive genes, those that perform basic “housekeeping” functions and are nearly always active at low levels, require only a basic set of regulatory controls; whereas those that that are activated by environmental signals, known as inducible genes, have a more specialized architecture.
This finding in yeast could open the door to a better understanding of the regulatory architecture of the human genome.
A paper describing the research by Penn State and Cornell University scientists appears March 10 in the journal Nature.
“When I first learned about DNA, I was taught to think of the genome as a library containing every book ever written,” said Matthew J. Rossi, research assistant professor at Penn State and the first author of the paper. “The genome is stored as part of a complex of DNA, RNA and proteins, called ‘chromatin.’ The interactions of the proteins and DNA regulate when and where genes are expressed to produce RNA (i.e. reading a book to learn or make something specific). But what I always wondered was with all that complexity, how do you find the right book when you need it? That is the question we are trying to answer in this study.”
How a cell chooses the right book depends on regulatory proteins and their interaction with DNA in chromatin, what can be referred to as the regulatory architecture of the genome. Yeast cells can respond to changes in their environment by altering this regulatory architecture to turn different genes on or off. In multicellular organisms, like humans, the difference between muscle cells, neurons, and every other cell type is determined by regulating the set of genes those cells are expressing. Deciphering the mechanisms that control this differential gene expression is therefore vital for understanding responses to the environment, organismal development, and evolution.
“Proteins need to be recruited and assembled at genes for them to be switched ‘on,’” said B. Franklin Pugh, professor of molecular biology and genetics at Cornell University and a leader of the research project that was started when he was a professor at Penn State. “We’ve put together the most complete and high-resolution map of these proteins showing the locations that they bind to the yeast genome and revealing aspects of how they interact with each other to regulate gene expression.”
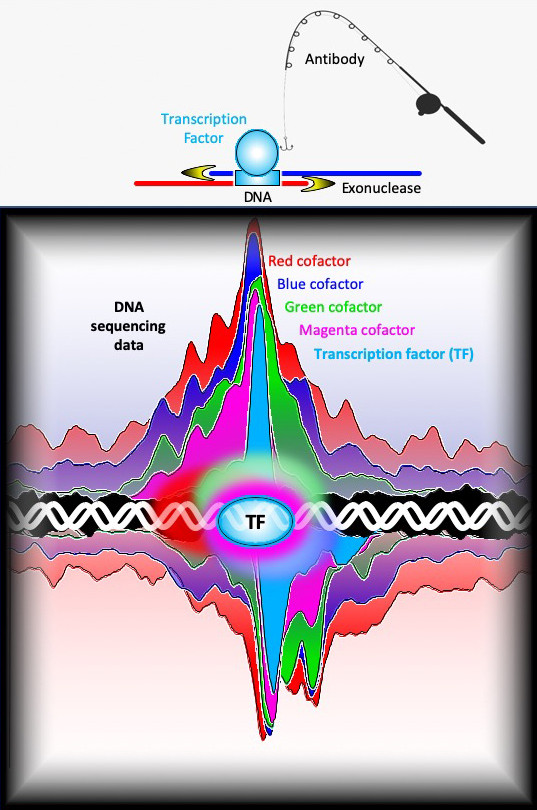
The team used a technique called ChIP-exo, a high-resolution version of ChIP-seq, to precisely and reproducibly map the binding locations of about 400 different proteins that interact with the yeast genome, some at a few locations and others at thousands of locations. In ChIP-exo, proteins are chemically cross-linked to the DNA inside living cells, thereby locking them into position. The chromosomes are then removed from cells and sheared into smaller pieces. Antibodies are used to capture specific proteins and the piece of DNA to which they are bound. The location of the protein-DNA interaction can then be found by sequencing the DNA attached to the protein and mapping the sequence back to the genome.
“In traditional ChIP-seq, the pieces of DNA attached to the proteins are still rather large and variable in length— ranging anywhere from 100 to 500 base pairs beyond the actual protein binding site,” said William K.M. Lai, assistant research professor at Cornell University and an author of the paper. “In ChIP-exo, we add an additional step of trimming the DNA with an enzyme called an exonuclease. This removes any excess DNA that is not protected by the cross-linked protein, allowing us to get a much more precise location for the binding event and to better visualize interactions among the proteins.”
The team performed over 1,200 individual ChIP-exo experiments producing billions of individual points of data. Analysis of the massive data leveraged Penn State’s supercomputing clusters and required the development of several novel bioinformatic tools, including a multifaceted computational workflow designed to identify patterns and reveal the organization of regulatory proteins in the yeast genome.
The analysis, which is akin to picking out repeated types of features on the ground from hundreds of satellite images, revealed a surprisingly small number of unique protein assemblages that are used repeatedly across the yeast genome.
“The resolution and completeness of the data allowed us to identify 21 protein assemblages and also to identify the absence of specific regulatory control signals at housekeeping genes,” said Shaun Mahony, assistant professor of biochemistry and molecular biology at Penn State and an author of the paper. “The computational methods that we’ve developed to analyze this data could serve as a jumping off point for further development for gene regulatory studies in more complex organisms.”
The traditional model of gene regulation involves proteins called ‘transcription factors’ that bind to specific DNA sequences to control the expression of a nearby gene. However, the researchers found that the majority of genes in yeast do not adhere to this model.
“We were surprised to find that housekeeping genes lacked a protein-DNA architecture that would allow specific transcription factors to bind, which is the hallmark of inducible genes,” said Pugh. “These genes just seem to need a general set of proteins that allow access to the DNA and its transcription without much need for regulation. Whether or not this pattern holds up in multicellular organisms like humans is yet to be seen. It’s a vastly more complex proposition, but like the sequencing of the yeast genome preceded the sequencing of the human genome, I’m sure we will eventually be able to see the regulatory architecture of the human genome at high resolution.”
In addition to Rossi, Pugh, Lai and Mahony, the research team includes Prashant K. Kuntala, Naomi Yamada, Nitika Badjatia, Chitvan Mittal, Guray Kuzu, Kylie Bocklund, Nina P. Farrell, Thomas R. Blanda, Joshua D. Mairose, Ann V. Basting, Katelyn S. Mistretta, David J. Rocco, Emily S. Perkinson and Gretta D. Kellogg.
This work was supported by the U.S. National Institutes of Health, the U.S. National Science Foundation, the Penn State Institute for Computational and Data Sciences, and Advanced CyberInfrastructure (ROAR) at Penn State.